Nonadiabatic Effects in the Oxygen Dissociation at Al(111)
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
The dissociation of oxygen molecules at metal surfaces is an important step in many technically important processes like heterogeneous catalysis and corrosion. In recent years, in particular the Al(111) surface has been frequently studied, because it has been considered as a relatively simple benchmark system for non-catalytically active metal surfaces.
Experimentally, it is well-known that the dissociative sticking probability of thermal oxygen molecules is very low [1-3]. Consequently, the existence of energy barriers for the dissociative adsorption has been postulated. In order to identify these barriers, several groups have studied the underlying potential-energy surface employing density-functional theory. Surprisingly, no sizable energy barriers could be identified [4,5], which could explain the experimental findings. Therefore, according to theory, the sticking probability should be essentially 100%, in contrast to experiment.
As a possible explanation it has been speculated that non-adiabatic effects might play an important role in the oxygen dissociation at Al(111) [4,6,7].
In order to address this problem, we have developed a constrained DFT method [8] in the spirit of Dederichs et al. [9] and implemented this method in the DMol3 code [10]. Employing this method we could identify significant energy barriers for the dissociative adsorption on the spin-triplet constrained energy surface [11,12]. This energy surface corresponds to the initial state of the system, when an oxygen molecule in its spin triplet ground state approaches the surface. If the transition probability to the final spin singlet energy surface of the adsorbed atoms is low, then these barriers might well account for the experimentally observed low sticking probability.
To confirm this, a large number of molecular dynamics trajectories had to be calculated to obtain statistically meaningful results. However, calculating thousands of trajectories by ab initio molecular dynamics on-the-fly is computationally too demanding. We have therefore constructed a Neural Network representation of the relevant energy surfaces based on several thousand constrained DFT energies for various molecular configurations [13]. This Neural Network representation then provides the energies and forces about five orders of magnitude faster than the underlying DFT calculations with about the same accuracy.
Employing this method we have calculated the sticking curve as a function of the kinetic energy of the oxygen molecules. A comparison with experimental data [3] from molecular beam experiments shows excellent agreement.
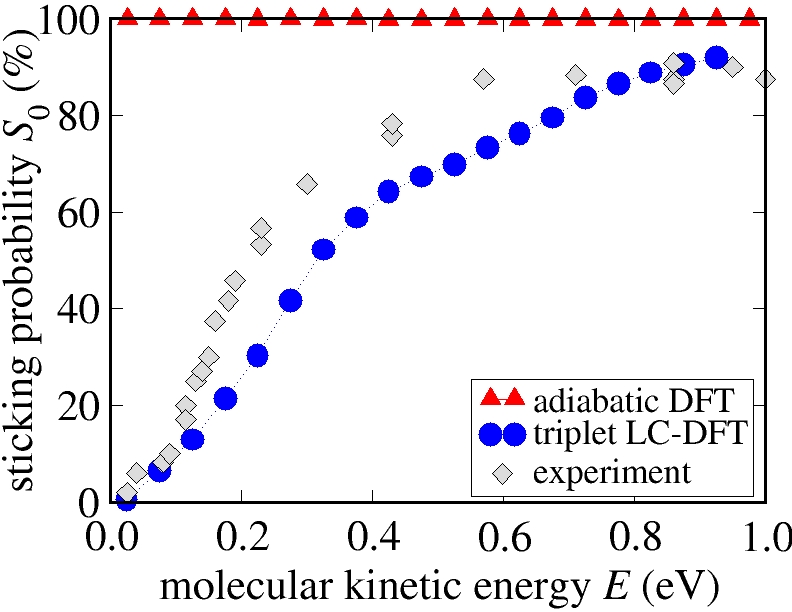
Sticking probability for oxygen molecules at the Al(111) surface.
Further, the simulations have been extended by including transitions from the initial triplet to the final singlet energy surface employing Tully's fewest switches algorithm [14,15].
[1] H. Brune, J. Wintterlin, R.J. Behm, and G. Ertl, Phys. Rev. Lett. 68 (1992) 624.
[2] A.J. Komrowski, J.Z. Sexton, A.C. Kummel, M. Binetti, O. Weisse, and E.Hasselbrink, Phys. Rev. Lett. 87 (2001) 246103.
[3] L. Oesterlund, I. Zoric, and B. Kasemo, Phys. Rev. B 55 (1997) 15452.
[4] K. Honkala, and K. Laasonen, Phys. Rev. Lett. 84 (2000) 705.
[5] Y. Yourdshahyan, B. Razaznejad , and B.I. Lundqvist, Solid State Comm. 117 (2001) 531.
[6] A. Hellman, B. Razaznejad, Y. Yourdshahyan, H. Ternow, I. Zoric, and B.I. Lundqvist, Surf. Sci. 532 (2003) 126.
[7] M. Binetti, O. Weisse, E. Hasselbrink, G. Katz, R. Kosloff, and Y. Zeiri, Chem. Phys. Lett. 373 (2003) 366.
[8] J. Behler, B. Delley, K. Reuter, and M. Scheffler, Phys. Rev. B 75 (2007) 115409.
[9] P.H. Dederichs, S. Bluegel, R. Zeller, and H. Akai, Phys. Rev. Lett. 53 (1984) 2512.
[10] B. Delley, J. Chem. Phys. 92 (1990) 508.
[11] J. Behler, B. Delley, S. Lorenz, K. Reuter, and M. Scheffler, Phys. Rev. Lett. 94 (2005) 36104.
[12] J. Behler, K. Reuter, and M. Scheffler, Phys. Rev. B 77 (2008) 115421.
[13] J. Behler, S. Lorenz, and K. Reuter, J. Chem. Phys. 127 (2007) 014705.
[14] C. Carbogno, J. Behler, A. Gross, and K. Reuter, Phys. Rev. Lett. 101 (2008) 096104.
[15] C. Carbogno, J. Behler, K. Reuter, and A. Gross, Phys. Rev. B 81 (2010) 035410.