A Neural Network Potential for R,R-Tartaric Acid
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
For the description of organic molecules a large number of force fields is available. However, most of these force fields are non-reactive and do not allow for the making and breaking of chemical bonds. In th e past decade, Neural Network potentials, which naturally incorporate this capability, have been employed to construct energy surfaces for a number of molecules (see [1,2] for reviews). In fact, these molecular potential energy surfaces have been the main applications of NN potentials for a long time. Nevertheless, the size of the molecules that could be addressed has been limited to a few atoms only due to technical difficulties in transforming the atomic positions to suitable input coordinates for the Neural Network.
In order to test the applicability of our high-dimensional Neural Network approach to molecular systems we have constructed a potential-energy surface for R,R-tartaric acid as a benchmark system.
As reference method for this study we have used the MM3 force field. The NN potential is able to reproduce accurately the energies of classical molecular dynamics trajectories at various temperatures. Based on these promising first results, we are currently replacing the force field by more accurate electronic structure calculations.
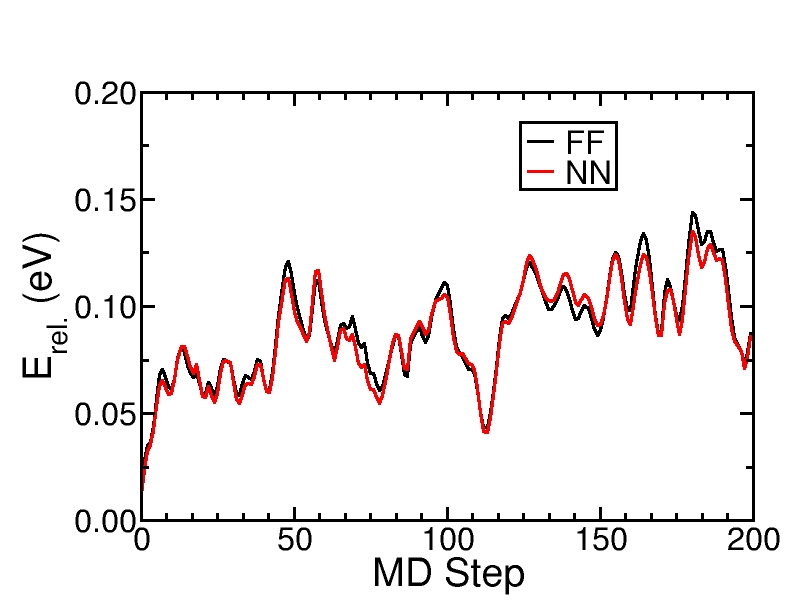
Comparison of the Neural Network (NN) and force field (FF) energies of a MD trajectory at 300 K.
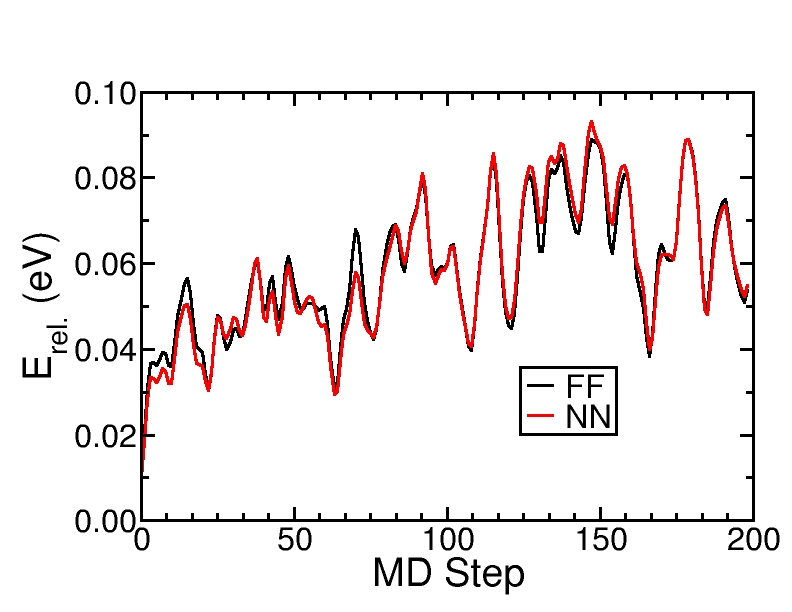
Comparison of the Neural Network (NN) and force field (FF) energies of a MD trajectory at 600 K.
[1] C. M. Handley, and P. L. A. Popelier, J. Phys. Chem. A 114 (2010) 3371.
[2] J. Behler, Chem. Modelling, in press (2010).