A Neural Network Potential for Water
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
In the context of the research focus
Solvation Science@RUB we have also started to construct a Neural Network potential for simulations of water.
Among all liquids water is the by far most frequently studied solvent due to its unique importance for living organisms.
Accordingly, a huge number of efficient water potentials of varying form and complexity can be found in the literature, ranging from very simple rigid molecules employing point charges to highly sophisticated
polarizable potentials.
Several suggestions have been made in recent years how to improve the theoretical description of water by conventional potentials employing Neural Networks [1-3].
Our high-dimensional Neural Network approach offers a natural description of flexible water molecules, which allows also for protonation and deprotonation of the molecules.
The potential is therefore capable of reproducing the energetics of ab initio molecular dynamics simulations very accurately.
In principle, the Neural Network potential can be constructed using any electronic structure reference method. We have chosen density functional theory (PBE functional). The following figure shows a comparison of the Neural Network and DFT energies of several structures obtained in a MD simulation.
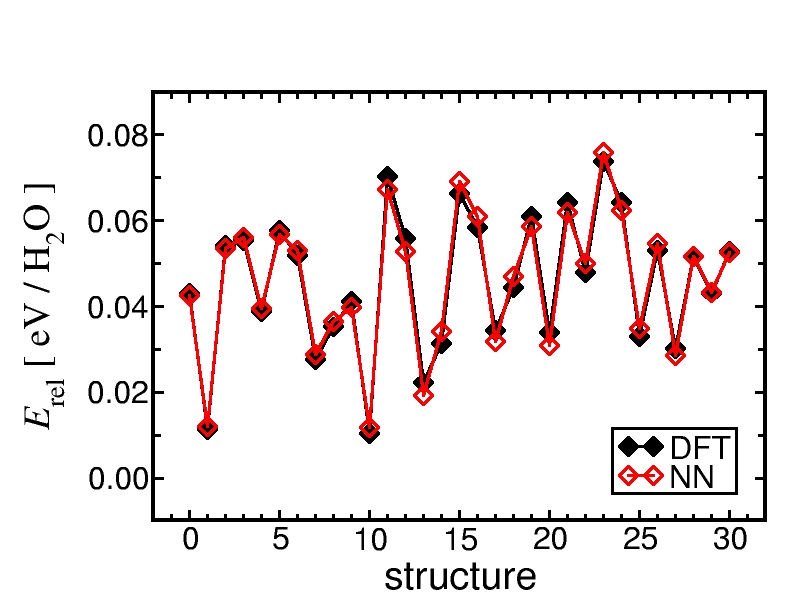
Comparison of the Neural Network and DFT energies for some bulk water structures extracted from a molecular dynamics simulation
[1] K. T. No, B. H. Chang, S. Y. Kim, M. S. Jhon, and H. A. Scheraga, Chem. Phys. Lett. 271, 152 (1997).
[2] K. W. Cho, K. T. No, and H. A. Scheraga, J. Mol. Struct. 641, 77 (2002).
[3] C. M. Handley, and P. L. A. Popelier, J. Chem. Theory Comp. 5, 1474 (2009).