A Neural Network Potential for Zinc Oxide
Lehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum
For multicomponent systems like oxides, charge transfer plays an important role. This charge transfer gives rise to long-range electrostatic interactions, which cannot always be neglected.
We have thus added an electrostatic energy term [1] to the high-dimensional NN method of Behler and Parrinello [2].
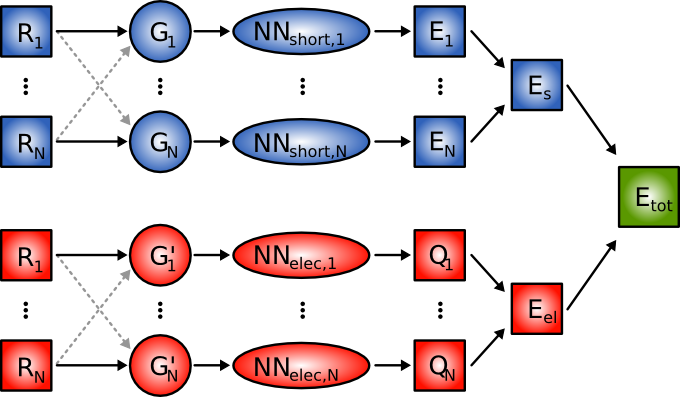
In the high-dimensional neural network(NN) method for multicomponent systems the total energy is obtained as a sum of a short-range energy Es,
which is a sum of atomic energies [2], and a long-range electrostatic energy Eel,
which is calculated from atomic charges QN. Both, the short-range atomic energies
and the atomic charges,depend on the local chemical environment of the atoms and are constructed by atomic NNs.
Here we illustrate this extension for the example of zinc oxide (ZnO).
ZnO is a technologically important material and is studied in the context of the collaborative research center "Metal-Substrate Interactions in Heterogeneous Catalysis"
(SFB558)
of the Ruhr-Universität Bochum.
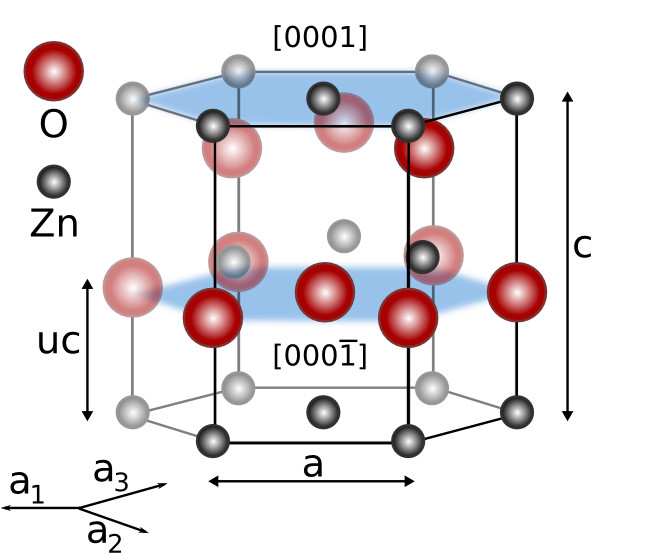
During the last decade the scientific interest in metal oxide
surfaces was steadily increasing. This is due to the great number of
applications that involve such materials ranging from gas sensors over
building blocks of optical devices and pigments in wall paints up to
-- most notably -- heterogeneous catalysis.
Zinc oxide, as an example, is a high temperature catalyst for the
methanol synthesis.
Although much effort has been spent to understand the atomic and
electronic properties of metal oxides, there is still a big lack of
knowledge because the systems in question are far more complex than
pure metals.
In order to study large systems in detail,
we have constructed a neural network potential for zinc oxide.
The structural and energetic properties of a wide range of zinc oxide structures e.g.
crystal structures, amorphous structures, clusters, and surfaces of zinc oxide,
predicted by the NN potential are in excellent agreement with reference density-functional theory (DFT)
calculations employing the FHI-aims code.[3]
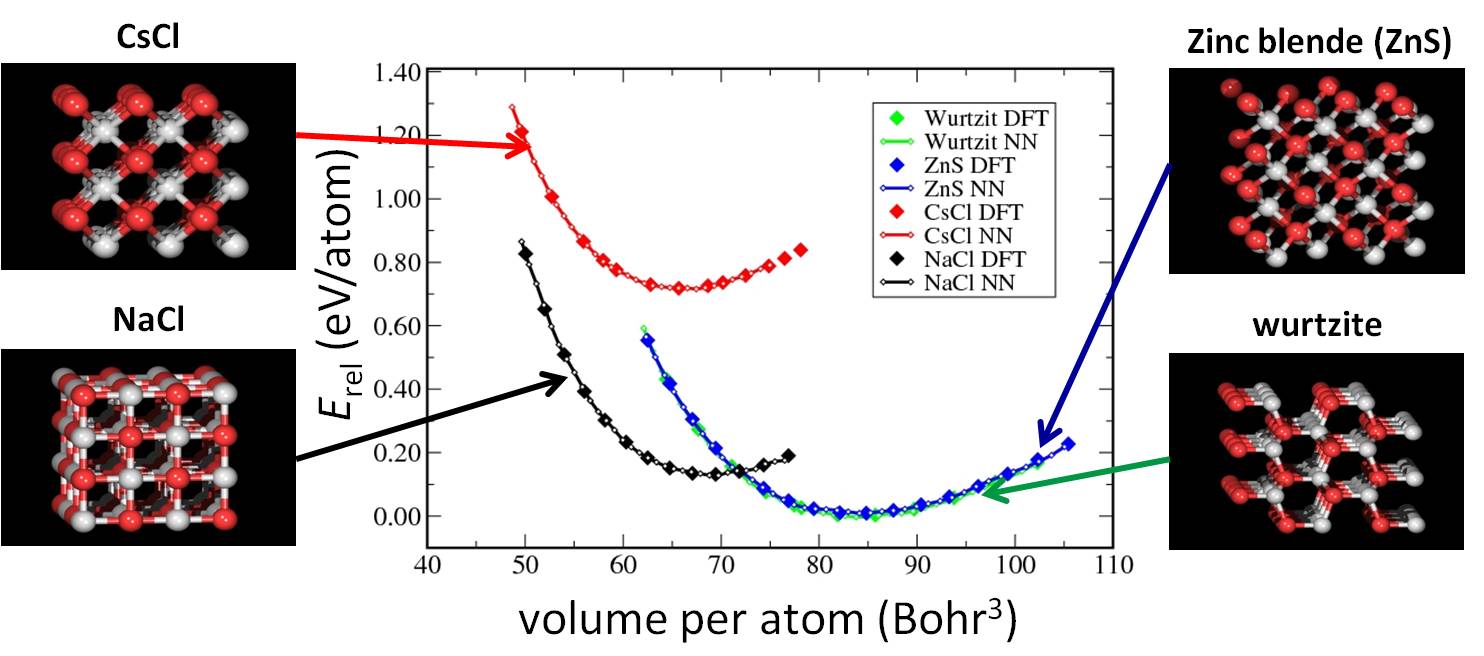
Energy vs. volume curves for several crystal structures of ZnO.
The solid diamonds represent DFT energies while the lines
correspond to the neural network (NN) potential-energy surface.
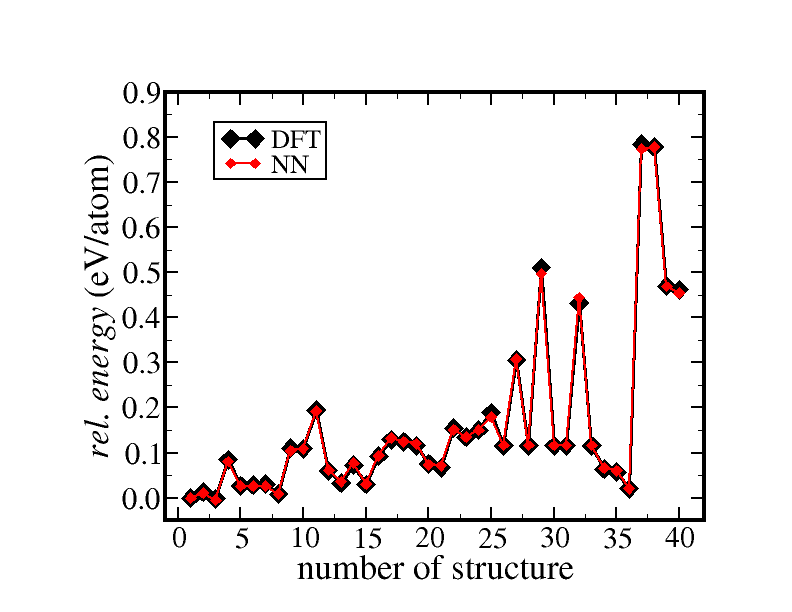
Comparison of the NN and DFT energies for different slab structures
References
[1] N. Artrith, T. Morawietz, and J. Behler, Phys. Rev. B 83, 153101 (2011).
[2] J. Behler, and M. Parrinello, Phys. Rev. Lett. 98, 146401 (2007).
[3] V. Blum et al., Comp. Phys. Comm. 180, 2175 (2009).
Back to the top of page