Although my current job leaves me too little time to pursue any regular research on my own, i nevertheless try hard to still keep a few research projects of my own going. I also try to stay involved with some of the ongoing research projects in our department, mostly connected to classical molecular dynamics simulations of aqueous systems, the field of my Diplom and Ph.D thesis, and the area of my largest scientific expertise. Yet after spending more than three years with a mean gang of hard-core quantum chemists and theoretical physicists i seem to pick up some expertise in their field as well.
The Impact of System Size on Properties of Bulk Water in Molecular Dynamics Simulations
 Usually the actual selection of some system parameters in molecular
dynamics (MD) simulations, for example the system size or the method to
treat the long-range Coulomb interactions, is more due to practical
reasons than physical criteria. Especially the amount of available
computer power (or lack thereof, to be more precise) or the (lack of)
capabilities of the employed simulation software enforce making compromises.
But since the stucture large aqueous systems is highly complex, it is
difficult to predict the impact of changing certain parameters.
So one would have to follow a try-and-err scheme until the simulation
'works', but that would mean to do several simulations.
Thus one generally tries to play it safe by running the largest possible
system, re-using parameters from other, similar simulations, or just
by following mixture of previous experience, intuition and instinct.
Usually the actual selection of some system parameters in molecular
dynamics (MD) simulations, for example the system size or the method to
treat the long-range Coulomb interactions, is more due to practical
reasons than physical criteria. Especially the amount of available
computer power (or lack thereof, to be more precise) or the (lack of)
capabilities of the employed simulation software enforce making compromises.
But since the stucture large aqueous systems is highly complex, it is
difficult to predict the impact of changing certain parameters.
So one would have to follow a try-and-err scheme until the simulation
'works', but that would mean to do several simulations.
Thus one generally tries to play it safe by running the largest possible
system, re-using parameters from other, similar simulations, or just
by following mixture of previous experience, intuition and instinct.
 The systematic examination of the impact of the system size on
static, dynamic and dielectric properties of bulk water systems can give a
valuable guideline for the selection of simulation parameters.
For a number of reasons the investigation of pure bulk water seems
useful:
The systematic examination of the impact of the system size on
static, dynamic and dielectric properties of bulk water systems can give a
valuable guideline for the selection of simulation parameters.
For a number of reasons the investigation of pure bulk water seems
useful:
- As a one component system it is easier to get good statistics and converged properties.
- The three-dimensional hydrogen bonded network of water molecules is a highly complex structure. In combination with the large dipole moment of water, dielectric properties of the bulk will very likely exhibit changes if system size and/or boundary conditions have a significant influence.
- The results are quite likely transferable to electrolytes and many other aqueous systems, as water is - by far - the main component.
- When using periodic boundary conditions, one has a maximally homogenous sample, so that any difference between simulations can be attributed to either system size of treatment of long-range interactions.
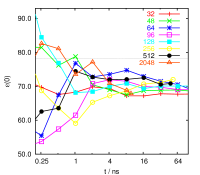
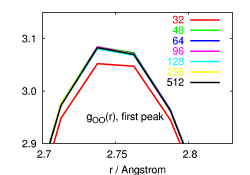
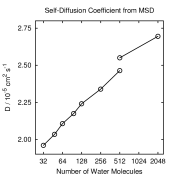
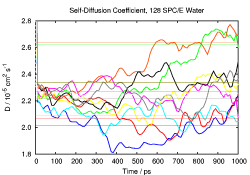 Properties to look at include pair correlation functions, self diffusion
coefficients, dipole auto-correlation functions, static
dielectric constants and size dependent Kirkwood G-factors. So far it
was found, that the influence of simulation times and system size are
quite different for different properties. Radial distribution functions
are reproduced well even from small systems and short trajectories (see
top left picture). Self-diffusion on the other hand is influenced by
both (see pictures on the right side), while the static dielectric
constant (left) is mainly influenced by the simulation time. For some
more detailed results, see e.g. the slides from a
presentation at ICTP in
Trieste in March 2004.
Properties to look at include pair correlation functions, self diffusion
coefficients, dipole auto-correlation functions, static
dielectric constants and size dependent Kirkwood G-factors. So far it
was found, that the influence of simulation times and system size are
quite different for different properties. Radial distribution functions
are reproduced well even from small systems and short trajectories (see
top left picture). Self-diffusion on the other hand is influenced by
both (see pictures on the right side), while the static dielectric
constant (left) is mainly influenced by the simulation time. For some
more detailed results, see e.g. the slides from a
presentation at ICTP in
Trieste in March 2004.
Chemical Visualization
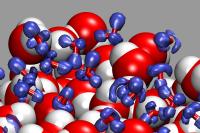
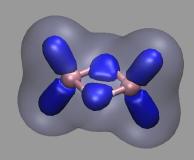 Results from quantum chemical calculations and computer simulations need
to be visualized adequately to make them easy to comprehend. There are
many visualization programs available, yet they can only provide some
standard representations for normal data. For more advanced
and instructive visualizations, especially from quantum chemical simulations
one needs to add some additional data processing, extend the visualization software,
adapt the simulation program, or all of the above. So far, this effort has yielded
a Visualization Tutorial,
contributions to scientific magazine covers (e.g.
Physik Journal, 5/2004
or
PCCP 8/2004), images for several articles in journals,
and some contributed code to the
Visual Molecular Dynamics (VMD)
program, the CPMD simulation program
and the ESPRESSO
electronic structure and first principles molecular dynamics program package.
Results from quantum chemical calculations and computer simulations need
to be visualized adequately to make them easy to comprehend. There are
many visualization programs available, yet they can only provide some
standard representations for normal data. For more advanced
and instructive visualizations, especially from quantum chemical simulations
one needs to add some additional data processing, extend the visualization software,
adapt the simulation program, or all of the above. So far, this effort has yielded
a Visualization Tutorial,
contributions to scientific magazine covers (e.g.
Physik Journal, 5/2004
or
PCCP 8/2004), images for several articles in journals,
and some contributed code to the
Visual Molecular Dynamics (VMD)
program, the CPMD simulation program
and the ESPRESSO
electronic structure and first principles molecular dynamics program package.
The Influence of Solvation and Hydrogen-Bonding on the Folding and Unfolding of Peptides.
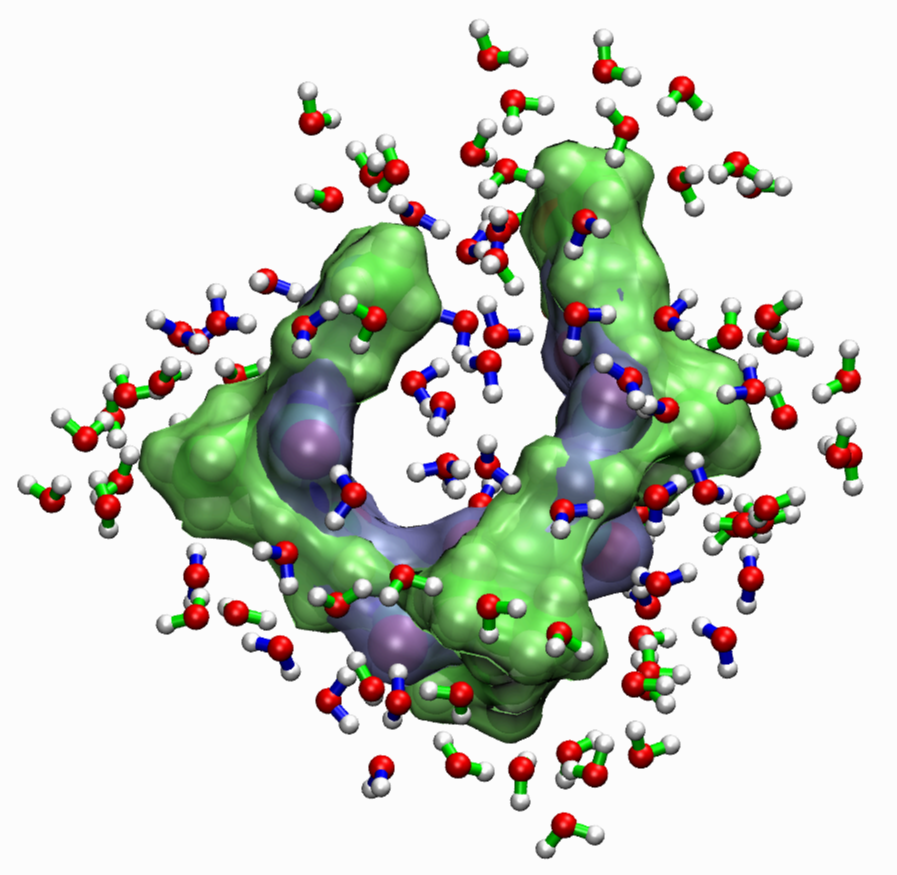 Proteins usually unfold or denaturate if heated well beyond
physiological conditions. Elastin proteins exhibit (to some extent) the
opposite behavior: they shrink (or fold) upon heating. A process which
has been named inverse temperature transition (ITT). It is widely
recognized that water molecules play an integral role in the stabilizing of the
(un-)folded state, the mechanism itself is still controversely
discussed. We want to gain further knowledge of the details of the
mechanisms using classical molecular dynamics simulations of the
smallest building blocks of elastin proteins and complementary
experimental studies of the same peptides.
Proteins usually unfold or denaturate if heated well beyond
physiological conditions. Elastin proteins exhibit (to some extent) the
opposite behavior: they shrink (or fold) upon heating. A process which
has been named inverse temperature transition (ITT). It is widely
recognized that water molecules play an integral role in the stabilizing of the
(un-)folded state, the mechanism itself is still controversely
discussed. We want to gain further knowledge of the details of the
mechanisms using classical molecular dynamics simulations of the
smallest building blocks of elastin proteins and complementary
experimental studies of the same peptides.
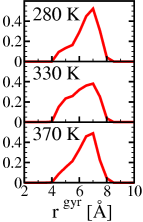 For the Octapeptide GVGVPGVG, the smallest of the Elastin like peptides,
the ITT could be verified experimentally which in turn made it possible
to run long enough (classical) molecular dynamics trajectories to do
some meaningful statistical analysis. As can be seen from the graph on the right,
there is an increased probability of a lower radius of gyration at 330K
comparison to the distribution at 280K and 370K
(see e.g.
Phys. Rev. Lett., 92 (14), 148101, (2004), and
Biophys. J,. 86,
1393-1407, (2004) for more details).
For the Octapeptide GVGVPGVG, the smallest of the Elastin like peptides,
the ITT could be verified experimentally which in turn made it possible
to run long enough (classical) molecular dynamics trajectories to do
some meaningful statistical analysis. As can be seen from the graph on the right,
there is an increased probability of a lower radius of gyration at 330K
comparison to the distribution at 280K and 370K
(see e.g.
Phys. Rev. Lett., 92 (14), 148101, (2004), and
Biophys. J,. 86,
1393-1407, (2004) for more details).
This project was and still is fostered by Dominik Marx and Roger Rousseau and has benefitted and still benefits from their contributions as well as those by Nicolaj Otte, Eduard Schreiner, and Marcel Baer during their undergraduate studies.
See also the project description (german) on the homepage of the DFG Forschergruppe 436.
Cystein as Surfactant for ZnS Nanoparticles in Water
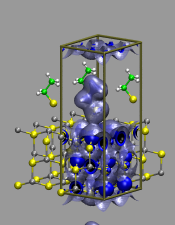
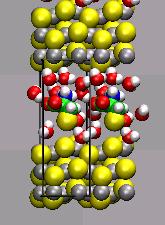 Mn-doped ZnS nanocrystals exhibit an orange fluorescence which makes
them useful, e.g. as indicator for medical imaging. With Cystein as
surfactant the unsoluble nanocrystals can be dispersed in water, but
there is little known about the way cystein couples to the ZnS suface. A deeper
knowledge of the binding mechanism could enable the use of ZnS as marker
for larger biomolecules.
Mn-doped ZnS nanocrystals exhibit an orange fluorescence which makes
them useful, e.g. as indicator for medical imaging. With Cystein as
surfactant the unsoluble nanocrystals can be dispersed in water, but
there is little known about the way cystein couples to the ZnS suface. A deeper
knowledge of the binding mechanism could enable the use of ZnS as marker
for larger biomolecules.
Also. the 'dangling bonds' of the uncoated ZnS-crystals lower the intensity
of the fluorescense significantly.
Periodic ab initio calculations and first-principles
Car-Parrinello molecular dynamics simulations are applied to study
selected properties of the nano-crystals and the surfactant binding
mechanism.
The Role of Proton Transfers on the Reaction Mechanisms of Acetylcholine Deacylation in AChE from Molecular Dynamics Simulations
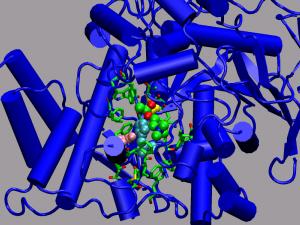 Acetylcholinesterase is a serine hydrolase that belongs to the esterase
family within higher eukaryotes. This family acts on different types of
carboxylic esters. AChE's biological role is the termination of the
nerve impulse transmissions at cholinergic synapses found at
neuromuscular junctions by decomposing acetylcholine into acetic acid
and choline. Since this process has to happen extremely fast this process
is a attractive target for molecular dynamics investigations (including
first principles MD), where sufficient statistical sampling is always a
problem for simulation of biomolecular processes. Key part to the
catalytic activity is a so-called catalytic triade for which a detailed
picture of its participation in the deacylation process has not (yet)
been found and thus several suggestions of the reaction mechanisms exist.
Acetylcholinesterase is a serine hydrolase that belongs to the esterase
family within higher eukaryotes. This family acts on different types of
carboxylic esters. AChE's biological role is the termination of the
nerve impulse transmissions at cholinergic synapses found at
neuromuscular junctions by decomposing acetylcholine into acetic acid
and choline. Since this process has to happen extremely fast this process
is a attractive target for molecular dynamics investigations (including
first principles MD), where sufficient statistical sampling is always a
problem for simulation of biomolecular processes. Key part to the
catalytic activity is a so-called catalytic triade for which a detailed
picture of its participation in the deacylation process has not (yet)
been found and thus several suggestions of the reaction mechanisms exist.
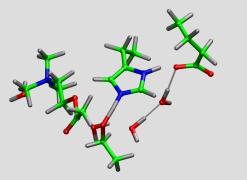 As a first step, a minimal model system (see picture) has
been investigated with a series of comparatively short Car-Parrinello
molecular dynamics simulations using ultrasoft pseudopotentials. These
tests have given proof, that a first priciples molecular dynamics is an
efficient tool for this kind of system and some of the steps in the
reaction mechanism do happen spontaneously, even at the time scale of a
few picoseconds. Also the influence of the solvent and the backbone
motions could be seen. For more details, have a look at the bachelor
thesis of Rachel Glaves, who did the bulk of these initial studies.
As a first step, a minimal model system (see picture) has
been investigated with a series of comparatively short Car-Parrinello
molecular dynamics simulations using ultrasoft pseudopotentials. These
tests have given proof, that a first priciples molecular dynamics is an
efficient tool for this kind of system and some of the steps in the
reaction mechanism do happen spontaneously, even at the time scale of a
few picoseconds. Also the influence of the solvent and the backbone
motions could be seen. For more details, have a look at the bachelor
thesis of Rachel Glaves, who did the bulk of these initial studies.
![]() B.Sc. thesis of R. Glaves (PDF/english)
B.Sc. thesis of R. Glaves (PDF/english)
Disclaimer / Author of this page: Axel.Kohlmeyer@theochem.ruhr-uni-bochum.de
Source File: research.wml (Mon Jan 10 18:54:49 2005) ($Revision: 1.14 $) Translated to HTML: Mon Oct 10 00:07:26 2005