Application of Car-Parrinello Molecular Dynamics Simulations:
Proton Transport in Bulk Water.
The following tutorial was adapted and translated from
 our website
with exercises in theoretical chemistry and biochemistry for
undergraduate students in chemistry and biochemistry.
Feel free to contact me at axel.kohlmeyer@theochem.ruhr-uni-bochum.de,
for any kind of feedback e.g. corrections, enhancements, or questions.
If you plan to use this material for your own courses,
please include a reference to the master website of our 'virtual lab',
http://www.theochem.ruhr-uni-bochum.de/go/lab.html.
our website
with exercises in theoretical chemistry and biochemistry for
undergraduate students in chemistry and biochemistry.
Feel free to contact me at axel.kohlmeyer@theochem.ruhr-uni-bochum.de,
for any kind of feedback e.g. corrections, enhancements, or questions.
If you plan to use this material for your own courses,
please include a reference to the master website of our 'virtual lab',
http://www.theochem.ruhr-uni-bochum.de/go/lab.html.
![]() Updated: 2004/02/06:
Updated: 2004/02/06:
- The (binary) pseudopotential files have been replaced with (text-mode) files that can be read on all platforms.
- The pseudotpotentials and plane wave cutoff have been changed to give (much) more accurate results at the expense of cpu time.
- The default temperature has been raised from 300 K to 400 K to get more proton transfers.
- Insight in the Capabilities of Car-Parrinello molecular dynamics simulations
- Basic knowledge in setting up and performing Car-Parrinello-simulations
- Comparison of classical MD with Born-Oppenheimer-MD and Car-Parrinello-MD
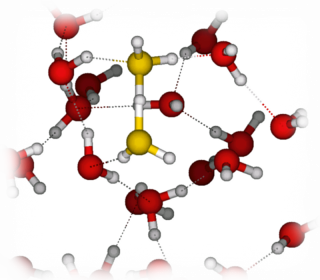
- Grotthuss-mechanism of proton transport in water
- generate a (equilibrated) start configuration
- calculate the wave-function for the start configuration
- perform the actual simulation
- analyze the trajectory
-
Create a classically equilibrated start configuration
Perform a classical molecular dynamics simulation of 32 water molecules (SPC/E potential) and 1 hydronium ion (adapted SPC/E potential) with the program Moldy. Use a cubic simulation cell with periodic boundary conditions, a density of 1 kg/l and a temperature of 400K. The simulation should cover at least 500 ps at a time step of 0.25. The equilibration works best, if you first run a 100 ps simulation at 700 K and use that restart to start the 400 K run.
The complete equilibration should take less than 8 hours on a 1.3 GHz AMD Athlon machine. Archive the coordinates and velocities of the last 50 ps of the trajectory at 2 fs distance for later analysis and/or visualization.
Downloads:
Moldy input file with potentials for 32 SPC/E water and 1 Hydronium ion.
For the impatient, there also is a already equilibrated restart. back to top
back to top
-
Calculation of the wave-function of the start configuration.
Convert the final configuration from the classical md-simulation into the xyz-format and use the xyz2cpmd.pl script to create the SYSTEM- and the ATOMS-section of the CPMD-input file. Add these sections to the CPMD input file template for a wave-function optimization and replace ##FIXME## with the edgelength (in angstrom) of the (cubic) unit cell of the classical simulation (= 9.95991 for the example configuration given above).
Similar to classical MD-simulations, where you have to provide model potentials (e.g. SPC/E for water), you now have to select an electron structure method. For the simulation of water the BLYP functional is recognized as a good choice.
Additionally you need pseudo-potential files for hydrogen (H_VDB_BLYP.psp) and oxygen (O_VDB_BLYP.psp) to start your first CPMD run. Finally you need to specify the size of the basis set. In case of plane waves you give the highest allowed fourier component, selected via the energy cutoff. For the pseudo-potentials given above, a cutoff of 25 Rydberg is recommended.
Now you can start the calculation with:./cpmd.x cpmd-init.wave.in > cpmd-init-wave.out
This run will need about 250MB of memory and take about 6 minutes on an Athlon 64 3200+ single processor machine to finish (see the reference output file for what it should look like. The reference run was done with CPMD version 3.9 and will give slightly different energy values on other platforms or with other CPMD versions).
back to top
-
Relaxation of the start configuration.
Modify the input file by replacing OPTIMIZE WAVEFUNCTION with
MOLECULAR DYNAMICS CP
RESTART WAVEFUNCTION COORDINATES LATEST
TEMPCONTROL IONS
400.0 50.0
MAXSTEP
200
TRAJECTORY SAMPLE
20
TIMESTEP
4.0
EMASS
600.0That starts - perusing the already optimized wave-function - a short (~20 fs, takes about 1 hour on an Athlon 64 3200+ cpu) Car-Parrinello simulation at 400 K. Temperature is controlled by rescaling of the atomic velocities, if the temperature differs more than 50 K from the configured value (see the reference output file for what it should look like). Now you should re-optimize the wavefunction and continue the simulation for 2-3 more short runs. This is most conveniently done by replacing the RESTART line in the input file with
RESTART WAVEFUNCTION COORDINATES VELOCITIES ACCUMULATORS LATEST
QUENCH BOELECTRONSAs the system relaxes you should be able to reduce the convergence parameter in small steps to 1*10-6. The 'quench to the Born-Oppenheimer surface' should not take more than 50 steps. Otherwise stop the simulation (with 'touch EXIT', cf. manual) and adjust the convergence parameter. Also, monitor the fictitious kinectic energy (EKINC). For the given parameters, it should stay around 0.02 a.u. If it becomes much larger, then you need to reoptimize the wavefunction, as you system is about to deviate too far from the Born-Oppenheimer Surface.
Discuss the progression of the several energies during the simulation (see file ENERGIES).
back to top
-
Simulation of a trajectory.
With the thusly prepared restart you can now run a full production simulation. Temperature control is now done with a Nosé-Hoover algorithm, which keeps the system during the simulation in the so called canonical ensemble (nVT-ensemble, see Car-Parrinello Review). You can do one of the following simulations:
- Car-Parrinello Simulation at 400 K. Replace:
TEMPCONTROL IONS
with
400.0 50.0
MAXSTEP
200TEMPERATURE
400.0
NOSE IONS MASSIVE
400.0 2500.0
NOSE ELECTRONS
0.02 10000.0 - Car-Parrinello Simulation at 300 K. Replace:
TEMPCONTROL IONS
with
400.0 50.0
MAXSTEP
200TEMPERATURE
400.0
NOSE IONS MASSIVE
300.0 2500.0
NOSE ELECTRONS
0.02 10000.0 - Born-Oppenheimer Simulation at 400 K. Replace:
MOLECULAR DYNAMICS CP
with
TEMPCONTROL IONS
400.0 50.0
MAXSTEP
200MOLECULAR DYNAMICS BO
TEMPERATURE
400.0
NOSE IONS MASSIVE
400.0 2500.0 - Car-Parrinello Simulation at 400 K with heavy
water and a (somewhat) larger time step. Replace:
TEMPCONTROL IONS
with
400.0 50.0
MAXSTEP
200TEMPERATURE
Add the following text to the section &ATOMS
400.0
NOSE IONS MASSIVE
400.0 2500.0
NOSE ELECTRONS
0.02 10000.0ISOTOPES
and change the TIMESTEP parameter to 6.0 a.u.
16.0
2.0
back to top
- Car-Parrinello Simulation at 400 K. Replace:
-
Visualization.
Use the script traj2xyz.pl to convert the trajectory to an xmol-style xyz-movie file. You can look at the trajectory with programs like Molden or VMD.
Use the script highlight.pl to highlight the hydronium ion in the xyz-file (der oxygen will be renamed to nitrogen) and visualize the trajectory with Molden. Alternatively you can use one of the techniques described in the CPMD VMD tutorial. The file http://ftp.theochem.ruhr-uni-bochum.de/outgoing/axel_kohlmeyer/cpmd-pics/movie-grotth.mpg contains a short rendered MPEG movie of such a visualization.
Convert the trajectory of the classical md into dcd format and visualize it with VMD (see the Moldy manual, on how to do that. Compare what you see to the CPMD run(s) and discuss similarities and differences between the trajectories. What are the differences between the individual simulations. Rate the computational effort of all methods.
-
Analysis.
Calculate the oxygen-oxygen and hydrogen-hydrochen pair correlation functions and compare the results with the respective pair correlation functions from the different ab initio and classical MD simulations. Some (older) reference trajectories are available on our FTP-Server
back to top
-
Excerpt from a larger overview article on Car-Parrinello methodology.
The full article is availble at http://www.theochem.ruhr-uni-bochum.de/go/cprev.html. Printed versions are available on request. -
Introductory article from Spektrum der Wissenschaft (german):
Further original articles on the same topic:
Protonenwanderung im virtuellen Labor.
-
The nature of the hydrated excess proton in water
from Nature and the review of that article from the same journal. -
The nature and transport mechanism of hydrated hydroxide ions in aqueous solution again from Nature plus a short review from the same journal and a more elaborate discussion of the article from Angewandte Chemie.
-
Fast Anomalous Diffusion of Small Hydrophobic Species in Water
from Physical Review Letters.
-
-
Webpage "Water structure and behavior" von Martin Chaplin
-
Homepage of the Forschergruppe 436:
Polymorphismus, Dynamik und Funktion von Wasser an molekularen Grenzflächen -
Homepage of the CPMD consortium
Goals of this exercise
Introduction
Modern first priciples Car-Parrinello methods render it possible to simulate complex molecular systems, e.g. water, gold surfaces, proteins, DNA-bases, and to study processes like diffusion, catalysis, or photoreactions with atomistics detail. The interactions in such complex systems can not be easily described by fixed, parametrized potentials, like it is usually done for classical molecular dynamics (MD) simulations. In a Car-Parrinello MD simulation, however, the interactions are calculated directly from the electron structure in every time step. The forces are obtained from the gradients of the total electronic energy at the positions of the nuclei and thus forming a multi-particle potential.
The determination of the total energy is done in the framework of density functional theory (DFT) utilizing plane waves basis functions to represent the valence electrons and pseudo-potentials to describe the atomic cores including the inner electrons. This method allows for an efficient modelling of extended systems, especially crystals or bulk liquids (when using periodic boundary conditions). The peculiarity of the Car-Parrinello approach is, that the wave-function is propargated following a ficticious Newtonian dynamic while performing the MD simulation. Therefore the wave-function does not need to be recalculated in every simulation step and the computational effort is reduced significantly.
Performing a Car-Parrinello MD-simulation somewhat resembles classical MD simulations and is usually done along the following steps.
Tasks
Before starting any calculations, you should (re-)acquaint yourself with the Car-Parrinello methodology and the CPMD Manual. Please remember, that a running simulation does not automatically mean, that the simulation is correctly representing the intended system. Therefore you should carefully examine the outputs of the simulations. This is especially important for the CPMD runs, since they are extremely time consuming. So you should carefully plan how to schedule the individual simulations.
Additional Information
Disclaimer / Author of this page: Axel.Kohlmeyer@theochem.ruhr-uni-bochum.de
Source File: cpmd-intro.wml (Tue May 10 22:43:35 2005) ($Revision: 1.13 $) Translated to HTML: Mon Oct 10 00:07:29 2005