
 Fig. 1:
Fig. 1:
![]()
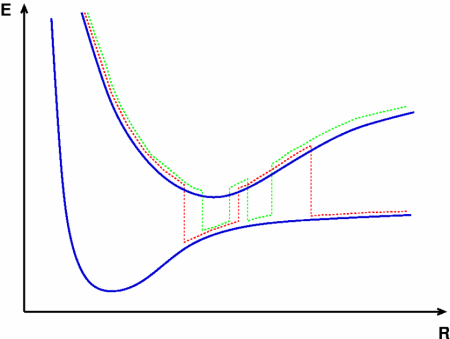
Chemical reactions rarely take place spontaneously; energy generally needs to be supplied in order to activate them. In the case of thermal reactions an activation barrier in the electronic ground state is overcome by heating the system. Photoreactions, on the other hand, only occur upon absorption of light promoting an electron into an excited state. Often the excited state dynamics is not entirely independent of the ground state (or other excited states), i.e. the two (or more) electronic states are coupled. In such a scenario one speaks of nonadiabatic dynamics.
Theoretical treatment of nonadiabatic dynamics requires two main ingredients, a reliable method to calculate excited states and a technique to perform molecular dynamics (MD) simulations in two or more coupled electronic states. Car-Parrinello (or ab initio) molecular dynamics (CP-MD) is a technique to simultaneously propagate atomic nuclei and their electrons based on density functional theory (DFT). Until recently this method was applicable only to systems in the electronic ground state. In 1998, a novel CP-MD scheme was introduced that made possible MD simulations in the lowest excited singlet state, S1. We have combined the ground and excited state CP-MD approaches and, in addition, incorporated nonadiabatic effects by implementing a so-called surface hopping algorithm (see Fig. 1). Our nonadiabatic CP-MD (na-CP-MD) method has been tested on a well-known photochemical reaction in the gas phase, namely the cis-trans photoisomerisation of formaldimine (see Fig. 2). Photoisomerisation about a double bond plays an important role in many biological processes such as vision. However, since real-life biochemical reactions usually take place in aqueous solution, it is desirable to be able to carry out nonadiabatic MD simulations of condensed phases. This is where the present na-CP-MD approach is most efficient. In order to demonstrate the applicability of na-CP-MD to condensed phase systems, we have studied the trans-cis photoisomerisation of diazene, N2H2, in aqueous solution (see Fig.3).
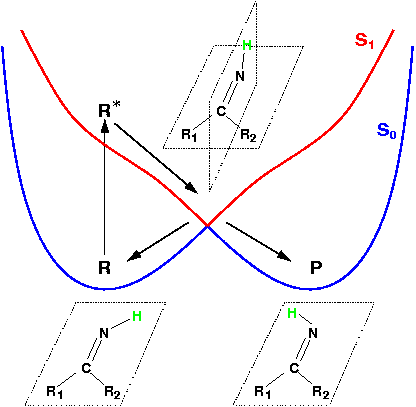 Fig. 2:
Fig. 2:![]()
Start animations above and below—about 4.0MByte
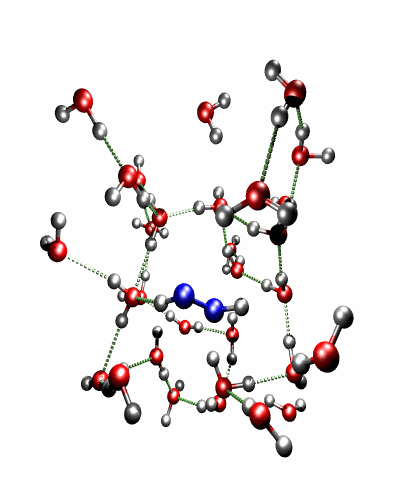
Fig. 3:
 Further information and details:
Further information and details:
- Nonadiabatic Car-Parrinello simulations
- Nikos Doltsinis (Ruhr-Universität Bochum)
in collaboration with - Dominik Marx (Ruhr-Universität Bochum)
- Nikos Doltsinis (Ruhr-Universität Bochum)
- Scientific visualization
- Nikos Doltsinis (Ruhr-Universität Bochum)
- Additional information
- RUB Pressemitteilung
- Nonadiabatic Car-Parrinello Molecular Dynamics method and formaldimine simulation:
N. L. Doltsinis and D. Marx,
Nonadiabatic Car-Parrinello Molecular Dynamics,
Phys. Rev. Lett. 88, 166402 (2002). - Review article on nonadiabatic ab initio simulations:
N. L. Doltsinis and D. Marx,
First Principles Molecular Dynamics Involving Excited States and Nonadiabatic Transitions,
J. Theor. Comput. Chem. 1, 319–349 (2002). - Expert introduction to nonadiabatic dynamics:
N. L. Doltsinis,
Nonadiabatic Dynamics: Mean Field and Surface Hopping,
in Quantum Simulations of Complex Many-Body Systems: From Theory to Algorithms,
edited by J. Grotendorst, D. Marx, and A. Muramatsu (NIC, FZ Jülich, 2002).
- This web page
http://www.theochem.ruhr-uni-bochum.de/go/surfhop.html