|
|
|
|
CPMD Tutorial Part 8 |
|
9. Exercise: Determination of Dynamic Properties
![]()
9.1. Calculation of Vibrational Spectra
![]()
9.2. The 'Dragging Effect'
![]()
![]() Part 7
Part 7
![]() Part 9
Part 9
![]() Start
Start
![]() Contents
Contents
 ... to be added.
... to be added.
... new reference runs needed. not yet revised.
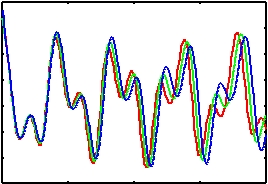
We now want to to have a look at the consequences of the 'dragging effect' of the fictitious dynamics during a CP-MD run. For this we look at 3 pre-calculated trajectories (1-2ps) of a single isolated water molecule:
 You can look up the inputs and outputs of the simulation in the
reference data section. If you look at the evolution of the various
energies during the simulation
(ENERGIES-bomd,
ENERGIES-cp-200au,
ENERGIES-cp-400au),
you get the impression, that all
trajectories seem to cover almost the same phase space, if the
initial kinetic energy added to the system takes into account
the extra amount needed for the fictitious dynamics of
the electronic system (which has been determined empirically in this case).
You can look up the inputs and outputs of the simulation in the
reference data section. If you look at the evolution of the various
energies during the simulation
(ENERGIES-bomd,
ENERGIES-cp-200au,
ENERGIES-cp-400au),
you get the impression, that all
trajectories seem to cover almost the same phase space, if the
initial kinetic energy added to the system takes into account
the extra amount needed for the fictitious dynamics of
the electronic system (which has been determined empirically in this case).
During the simulation also an analysis of the current dipole moment was performed and recorded (DIPOLE-bomd, DIPOLE-cp-200au, DIPOLE-cp-400au). These file can be used to calculate the infrared spectral densities using the provided fourier transform program . This code also adds several (optional) corrections to the spectra. It is recommended to look at the data in the first and the fifth column. You can see, that there is a noticeable red shift for some peaks in the spectral densities. The shift itself depends on the fictitious mass and on the mode (which makes it a bit tricky to compensate for it).
Full Table of Contents
![]()
1. Introduction
1.1. Development Notice
1.2. Notes
1.3. Recent Changes
1.4. Citation / Bookmark
2. Table of Contents
3. Preparation and Installation Issues
3.1. Compiling CPMD
3.2. Running CPMD
3.3. Running cpmd2cube
4. The Theory: Some Fundamental Infos and Useful Literature
5. The Basics: Running CPMD, Input and Output Formats
5.1. Wavefunction Optimization: a) Input File Format
5.2. Wavefunction Optimization: b) Output File Format
5.3. Geometry Optimization
5.4. Car-Parrinello Molecular Dynamics
5.5. Further Job Types
5.6. How to Use the Tutorial
6. Exercise: Electron Structure and Geometry Optimization
6.1. Hydrogen Molecule
6.2. Water Molecule
6.3. Ammonia Molecule
7. Exercise: Car-Parrinello Molecular Dynamics
7.1. Hydrogen Molecule
7.2. Ammonia Molecule in Gas Phase
7.3. Glycine Molecule in Gas Phase
7.4. Glycine with Thermostats
8. Exercise: Bulk Systems
8.1. Bulk Silicon
8.2. Hydronium Ion in Bulk Water
9. Exercise: Determination of Dynamic Properties
9.1. Calculation of Vibrational Spectra
9.2. The 'Dragging Effect'
10. Proton Transfer in a Catalytic Triade Model
10.1. Preparing a Model from a Large System
10.2. Equilibration with a Blocked Reaction Path
10.3. Modelling Part of the Reaction Path
10.4. Calculating Electron Structure Properties and Visualizations
11. Credits
12. Downloads
13. File distribution policy
|
|
|
||
|
|||