|
|
|
|
CPMD Tutorial Part 5 |
|
6. Exercise: Electron Structure and Geometry Optimization
![]()
6.1. Hydrogen Molecule
![]()
6.2. Water Molecule
![]()
6.3. Ammonia Molecule
![]()
![]() Part 4
Part 4
![]() Part 6
Part 6
![]() Start
Start
![]() Contents
Contents
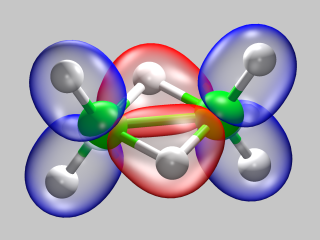
 The first object of study will be a rather trivial one:
an isolated hydrogen molecule. We will treat it in the Local Density
Approximation (LDA) and use a simple norm-conserving pseudopotential.
Although there are much better ways of treating this system, the calculations
are fast and the results easy to check, so that this is an ideal testbed to
introduce and try out a lot of features in CPMD.
The first object of study will be a rather trivial one:
an isolated hydrogen molecule. We will treat it in the Local Density
Approximation (LDA) and use a simple norm-conserving pseudopotential.
Although there are much better ways of treating this system, the calculations
are fast and the results easy to check, so that this is an ideal testbed to
introduce and try out a lot of features in CPMD.
Perform a wavefunction optimization (single point calculation) of a hydrogen molecule using the input file 1-h2-wave.inp and the pseudopotential file H_MT_LDA.psp. Rename the resulting restart file RESTART.1 to RESTART so that it can be used for the next calculations and will not be overwritten. Inspect the input and output files.
Calculate the Kohn-Sham Energies and calculate/write out several density or wavefunction type files using the RESTART file from the previous calculation (2-h2-ksener.inp). The resulting files DENSITY, ELPOT, etc. contain their information still in reciprocal space. You need to use the cpmd2cube.x utility to convert them to real space file in cube format. Usage, e.g.:
cpmd2cube.x -halfmesh -rho DENSITY cpmd2cube.x -halfmesh -psi WAVEFUNCTION.2 |
For the visualization of the resulting geometries and cube files, please see the VMD Visualization Tutorial. The visualization of cube files is described in Part 5. Plese note, that VMD does not consider bonds between two hydrogens (it makes it much more efficient for large biomolecules), so you have to manually set the bond with VMD scripting at the command prompt (or use a VDW representation):
set sel [atomselect 0 {name H}]
$sel setbonds {{1} {0}}
|
Again rename the RESTART.1 file to RESTART after you have verified, that the calculation was successful, so you can read it in the next step.
Calculate some more properties using the restart from the previous run. (3-h2-prop.inp). Have a look at the output file and the various other created files. See the CPMD manual for an explanation of their contents.
Try out various methods to optimize the wavefunction. The following input files are examples for the several methods implemented in CPMD. Compare them with respect to the final result and the convergence behavior:
Your can use the script comp-total-e.sh to extract a summary of the total energies from the single point calculations (1 + 4a-f).
Final question: how come you can use a pseudopotential for hydrogen, when there are no core electrons?
Requirements: Memory: 50-100 MB, CPU time: 1-5 min/job.
In the next section we will study a slightly more ambitious molecule: water. Since water has a dipole moment, you have to keep in mind, that we are calculating a system with periodic boundary conditions, so the water molecule 'sees' its images and interacts with them. There are methods implemented in CPMD to compensate for this effect, but we won't use them here to save resources. This time we will use a gradient corrected functional (BLYP) instead of the LDA. Also note that in the &ATOMS section the LMAX for the oxygen is set to P (instead of S for hydrogen) and that the keyword KLEINMAN-BYLANDER is required for for the calculation of the nonlocal parts of the pseudopotential.
Optimize the geometry of a water molecule with the default geometry optimization algorithm.
1-h2o-pbc-geoopt.inpRepeat the geometry optimization of a water molecule with the linear scaling geometry optimizer and adaptive convergence.
2-h2o-pbc-geo-linsc.inpDo a properties calculation using the RESTART from the previous run: 3-h2o-pbc-prop.inp
Requirements: Memory: 150-200 MB, CPU time: 2x 15-20 min + 1x 1 min.
In this example we will diversify a little more by looking at an ammonia molecule and using a different type of pseudopotential: a so-called 'ultra-soft' (Vanderbilt) pseudopotential (USPP). This class of pseudopotentials can be used with a much smaller plane wave cutoff and thus needs less memory resources than calculations with norm-conserving pseudopotentials. However, calculations with USPPs have to be set up more carefully and for the calculation of several properties a single-point wavefunction optimization with norm-conserving pseudopotentials has to be performed from the USPP restart. Also note that in this case the FORMATTED keyword is required to have the pseudopotential reader recognize the psedopotential file format.
Optimize the geometry of an ammonia molecule with ultra-soft
pseudopotentials:
1-nh3-geoopt-vdb.inp.
Note, that we now use SYMMETRY 0 and a
Poisson solver to decouple the periodic images, so that the result
is closer to the calculation of an isolated molecule. See the CPMD
manual for some further information.
Re-optimize the electronic structure with norm-conserving pseudopotentials
and calculate some data sets for visualization:
2-nh3-wfopt-mt.inp.
Note: this job needs much more memory, so you may
need to run it on a machine with more memory installed.
Perform a properties calculation based on the restart from the previous run. 3-nh3-prop-mt.inp.
Perform a geometry optimization of the (flat) transition state of the 'umbrella'-mode of the ammonia molecule with ultra-soft pseudopotentials: 4-nh3-geoopt-flat.inp. In this case we 'guide' the geometry optimizer to the desired structure by using a constraint, i.e. adding the following lines to the &ATOMS section of the input file.
CONSTRAINTS FIX STRUCTURE 1 TORSION 2 3 4 1 0.0 END CONSTRAINTS |
There also are geometry optimization algorithms implemented in CPMD that allow to search for a transition state, but this would go beyond the scope of this tutorial. Check out the CPMD manual and the CPMD test suite.
Requirements: Memory: 220 MB (VDB/25Ry), 600 MB (MT/70Ry), CPU time: 10-20 min (Properties: 1 min).
Full Table of Contents
![]()
1. Introduction
1.1. Development Notice
1.2. Notes
1.3. Recent Changes
1.4. Citation / Bookmark
2. Table of Contents
3. Preparation and Installation Issues
3.1. Compiling CPMD
3.2. Running CPMD
3.3. Running cpmd2cube
4. The Theory: Some Fundamental Infos and Useful Literature
5. The Basics: Running CPMD, Input and Output Formats
5.1. Wavefunction Optimization: a) Input File Format
5.2. Wavefunction Optimization: b) Output File Format
5.3. Geometry Optimization
5.4. Car-Parrinello Molecular Dynamics
5.5. Further Job Types
5.6. How to Use the Tutorial
6. Exercise: Electron Structure and Geometry Optimization
6.1. Hydrogen Molecule
6.2. Water Molecule
6.3. Ammonia Molecule
7. Exercise: Car-Parrinello Molecular Dynamics
7.1. Hydrogen Molecule
7.2. Ammonia Molecule in Gas Phase
7.3. Glycine Molecule in Gas Phase
7.4. Glycine with Thermostats
8. Exercise: Bulk Systems
8.1. Bulk Silicon
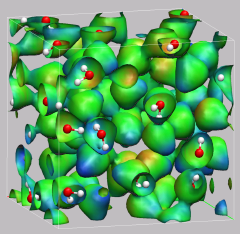
8.2. Hydronium Ion in Bulk Water
9. Exercise: Determination of Dynamic Properties
9.1. Calculation of Vibrational Spectra
9.2. The 'Dragging Effect'
10. Proton Transfer in a Catalytic Triade Model
10.1. Preparing a Model from a Large System
10.2. Equilibration with a Blocked Reaction Path
10.3. Modelling Part of the Reaction Path
10.4. Calculating Electron Structure Properties and Visualizations
11. Credits
12. Downloads
13. File distribution policy
|
|
|
||
|
|||