|
|
|
|
CPMD Tutorial Part 9 |
|
10. Proton Transfer in a Catalytic Triade Model
![]()
10.1. Preparing a Model from a Large System
![]()
10.2. Equilibration with a Blocked Reaction Path
![]()
10.3. Modelling Part of the Reaction Path
![]()
10.4. Calculating Electron Structure Properties and Visualizations
![]()
![]() Part 8
Part 8
![]() Part 10
Part 10
![]() Start
Start
![]() Contents
Contents
 ... new reference runs needed. not yet revised.
... new reference runs needed. not yet revised.
The final part of this tutorial shall demonstrate how to set up
a model system for a 'real-life' simulation. You can find
some background information about this model in this extract from the
Bachelor Thesis of Rachel Glaves.
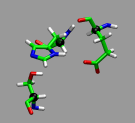
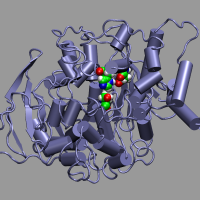 We want to look at the active sites in an
acetylcholineesterase enzyme.
The full enzyme, especially when including the solvating water molecules,
is by far too large to be treated with Car-Parrinello MD. Thus
the fist step is to extract a model system from a fully
solvated and equilibrated peptide. The main idea is to extract
a number of residues, only keep the amino acid sidechains and
the alpha-carbons which are turned in to methy groups and fixed in space.
To do this manually can become quite cumbersome, but the scripting
capabilities of VMD will help us to achieve our goal. The file
AChE.gro contains
the solvated and equilibrated enzyme. The VMD script
get-triade.vmd
will now extract the coordinates of the three residues that
comprise the triade and write them to a PDB file
(triade.pdb).
To run this script you can either start first start VMD and then load
the script via
source get-triade.vmd
from the VMD
terminal prompt or have VMD execute the script on loading via
vmd -e get-triade.vmd
. Since we don't need
the graphical user interface for pure script processing, you can
also use the
-dispdev none
option to VMD to disable
the GUI and the OpenGL window. The resulting PDB file will also
contain box size information that will add a 2 Angstrom safety margin
to avoid overlapping atoms via the periodic boundaries. Change the
script to extract only the HIS440 residue and run a geometry
optimization for the positions of the hydrogens only with CPMD
(with ultra-soft pseudopotentials and using FIX ATOMS).
We want to look at the active sites in an
acetylcholineesterase enzyme.
The full enzyme, especially when including the solvating water molecules,
is by far too large to be treated with Car-Parrinello MD. Thus
the fist step is to extract a model system from a fully
solvated and equilibrated peptide. The main idea is to extract
a number of residues, only keep the amino acid sidechains and
the alpha-carbons which are turned in to methy groups and fixed in space.
To do this manually can become quite cumbersome, but the scripting
capabilities of VMD will help us to achieve our goal. The file
AChE.gro contains
the solvated and equilibrated enzyme. The VMD script
get-triade.vmd
will now extract the coordinates of the three residues that
comprise the triade and write them to a PDB file
(triade.pdb).
To run this script you can either start first start VMD and then load
the script via
source get-triade.vmd
from the VMD
terminal prompt or have VMD execute the script on loading via
vmd -e get-triade.vmd
. Since we don't need
the graphical user interface for pure script processing, you can
also use the
-dispdev none
option to VMD to disable
the GUI and the OpenGL window. The resulting PDB file will also
contain box size information that will add a 2 Angstrom safety margin
to avoid overlapping atoms via the periodic boundaries. Change the
script to extract only the HIS440 residue and run a geometry
optimization for the positions of the hydrogens only with CPMD
(with ultra-soft pseudopotentials and using FIX ATOMS).
Requirements: Memory: 100 MB, CPU time: 10 min.
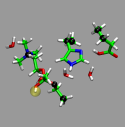 For the following steps we use the minimal triade subsystem with
the acetlycholine added (and bound) and including 4 water molecules.
Since we are looking at an
intermediate state of the enzymatic reaction, yet we have to equilibrate
the model system, as we have removed most of the
atoms surrounding the active site. So we have to find a way to inhibit
the reaction. In this case adding a proton and turning the ester group
into an semi-acetal group does the trick nicely. Use
1-prot-triade-wfnopt.inp
to calculate the optimized wavefunction and then start an MD run in the
usual way (2-prot-triade-equilib.inp).
In this case, we run only a few steps and skip the further equilibration
(3-prot-triade-cont.inp).
In fact, the start configuration was taken from a thusly equilibrated
configuration.
An xyz-movie file (TRAJEC.xyz)
of such an equilibration run is available.
For the following steps we use the minimal triade subsystem with
the acetlycholine added (and bound) and including 4 water molecules.
Since we are looking at an
intermediate state of the enzymatic reaction, yet we have to equilibrate
the model system, as we have removed most of the
atoms surrounding the active site. So we have to find a way to inhibit
the reaction. In this case adding a proton and turning the ester group
into an semi-acetal group does the trick nicely. Use
1-prot-triade-wfnopt.inp
to calculate the optimized wavefunction and then start an MD run in the
usual way (2-prot-triade-equilib.inp).
In this case, we run only a few steps and skip the further equilibration
(3-prot-triade-cont.inp).
In fact, the start configuration was taken from a thusly equilibrated
configuration.
An xyz-movie file (TRAJEC.xyz)
of such an equilibration run is available.
Requirements: Memory: 400 MB, CPU time: 20+15 min.
Now remove the inhibiting proton from the previous wavefunction optimization input, don't forget to adjust the rest of the input (CHARGE!) and start the 'real thing'. Equilibrate with (TEMPCONTROL IONS 300.0 20.0) for 60 steps and then switch to Nose-Hoover chains with the following parameters:
NOSE IONS MASSIVE 300.0 2500.0 NOSE ELECTRONS 0.007 15000.0 NOSE PARAMETERS 3 3 3 6.0D0 15 4 |
Now we need run the simulation at least until the next day to see a reaction to happen. Depending on the circumstances it will be either the forward or backward reaction. If you use the MAXCPU keyword you can set MAXSTEP as high as you want, after the indicated period the simulation will stop and write a restart. For instance with a value of 60000 for MAXCPU the job should stop after about 17 hours. This can also be extremely useful for running in a batch environment (especially in combinations with the RESTFILE and STORE keywords).
Requirements: Memory: 450 MB, CPU time: 45+60 min + 17h Production.
Since we were using ultra-soft pseudopotentials, we have to recalculate the electron structure with norm-conserving pseudopotentials (at least for most of the properties). Since this will need a lot of memory and CPU resources, already pre-calculated files are provided in the refout directory.
Requirements: Memory: 1200 MB, CPU time: 3-5 h/WFopt.
Full Table of Contents
![]()
1. Introduction
1.1. Development Notice
1.2. Notes
1.3. Recent Changes
1.4. Citation / Bookmark
2. Table of Contents
3. Preparation and Installation Issues
3.1. Compiling CPMD
3.2. Running CPMD
3.3. Running cpmd2cube
4. The Theory: Some Fundamental Infos and Useful Literature
5. The Basics: Running CPMD, Input and Output Formats
5.1. Wavefunction Optimization: a) Input File Format
5.2. Wavefunction Optimization: b) Output File Format
5.3. Geometry Optimization
5.4. Car-Parrinello Molecular Dynamics
5.5. Further Job Types
5.6. How to Use the Tutorial
6. Exercise: Electron Structure and Geometry Optimization
6.1. Hydrogen Molecule
6.2. Water Molecule
6.3. Ammonia Molecule
7. Exercise: Car-Parrinello Molecular Dynamics
7.1. Hydrogen Molecule
7.2. Ammonia Molecule in Gas Phase
7.3. Glycine Molecule in Gas Phase
7.4. Glycine with Thermostats
8. Exercise: Bulk Systems
8.1. Bulk Silicon
8.2. Hydronium Ion in Bulk Water
9. Exercise: Determination of Dynamic Properties
9.1. Calculation of Vibrational Spectra
9.2. The 'Dragging Effect'
10. Proton Transfer in a Catalytic Triade Model
10.1. Preparing a Model from a Large System
10.2. Equilibration with a Blocked Reaction Path
10.3. Modelling Part of the Reaction Path
10.4. Calculating Electron Structure Properties and Visualizations
11. Credits
12. Downloads
13. File distribution policy
|
|
|
||
|
|||